


Redacción. Las enfermedades mitocondriales son un grupo heterogéneo de trastornos genéticos raros causados por mutaciones en el ADN nuclear o mitocondrial (ADNmt). Estas enfermedades son frecuentemente multisistémicas, aunque afectan principalmente a tejidos que requieren grandes cantidades de energía como el cerebro. Las mutaciones en el ARN de transferencia mitocondrial (mt-tRNA) conducen a defectos en la traducción de proteínas que pueden comprometer algunas o todas las proteínas codificadas por el mtDNA. El síndrome de encefalomiopatía mitocondrial, acidosis láctica y episodios similares a accidentes cerebrovasculares (MELAS) está causado principalmente por la mutación m.3243A>G en el gen mt-tRNALeu(UUR) (MT-TL1). Actualmente, no existen tratamientos curativos para el síndrome MELAS por lo que es necesario el desarrollo de modelos de la enfermedad que permitan la búsqueda de terapias efectivas.
Debido a la falta de modelos animales adecuados, se han desarrollado varios modelos celulares para estudiar la enfermedad, proporcionando información sobre los mecanismos fisiopatológicos de MELAS.

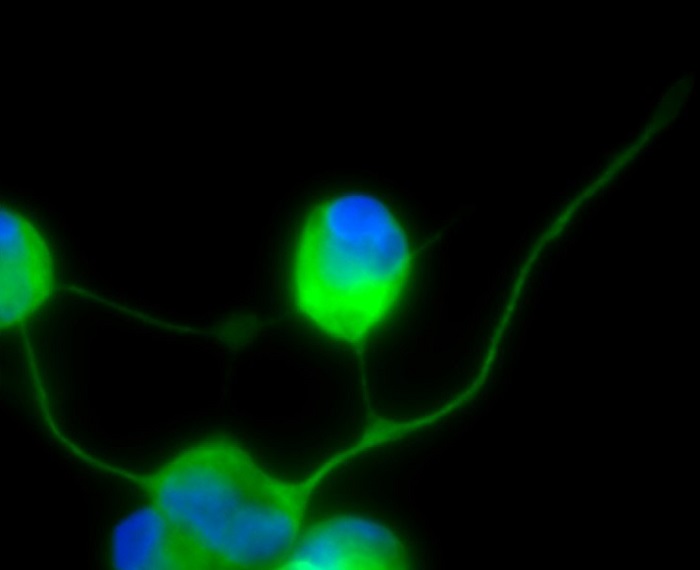
En este contexto, el equipo científico dirigido por el profesor de la Universidad Pablo de Olavide José Antonio Sánchez Alcázar, en colaboración con los profesores, también de la UPO, Antonio Rodríguez Moreno y José Ángel Armengol, han publicado recientemente un estudio que muestra una conversión directa exitosa de fibroblastos derivados de pacientes MELAS en neuronas inducidas.
Las neuronas generadas mantienen la mutación presente en las células de la piel fibroblastos y, cocultivados con astrocitos (células de soporte que proporcionan nutrición, apoyo y protección a las neuronas), muestran propiedades electrofisiológicas.






